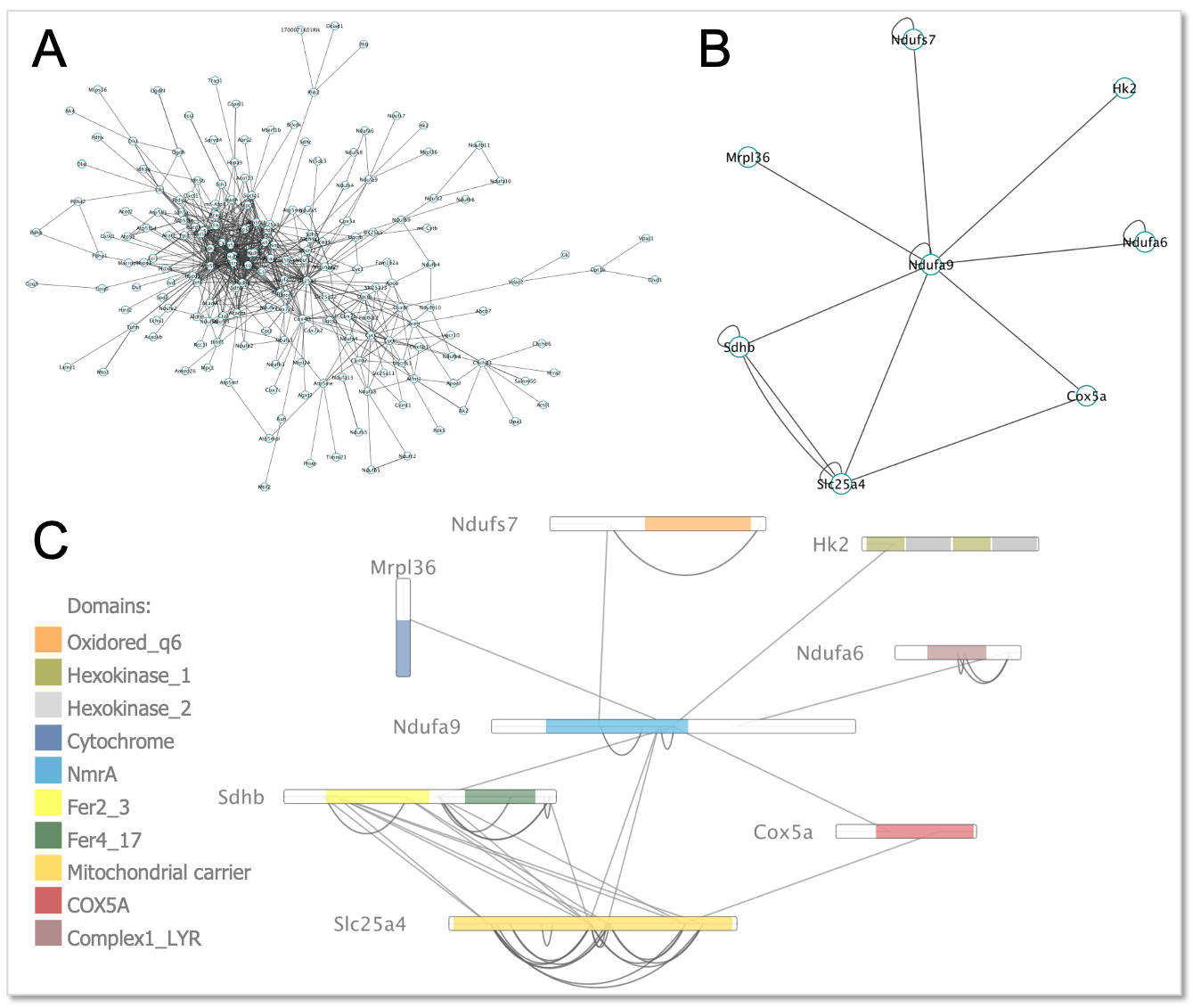
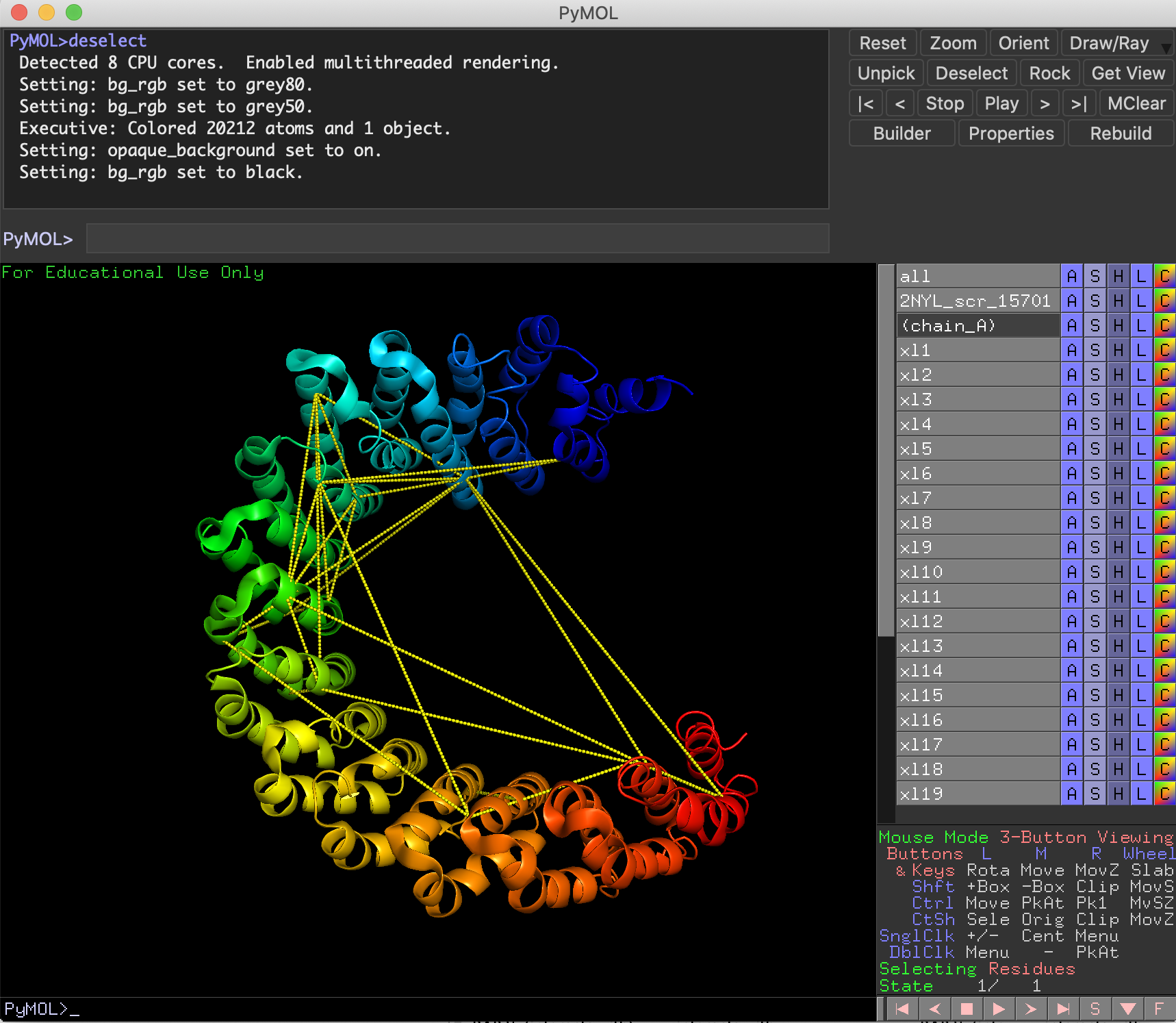
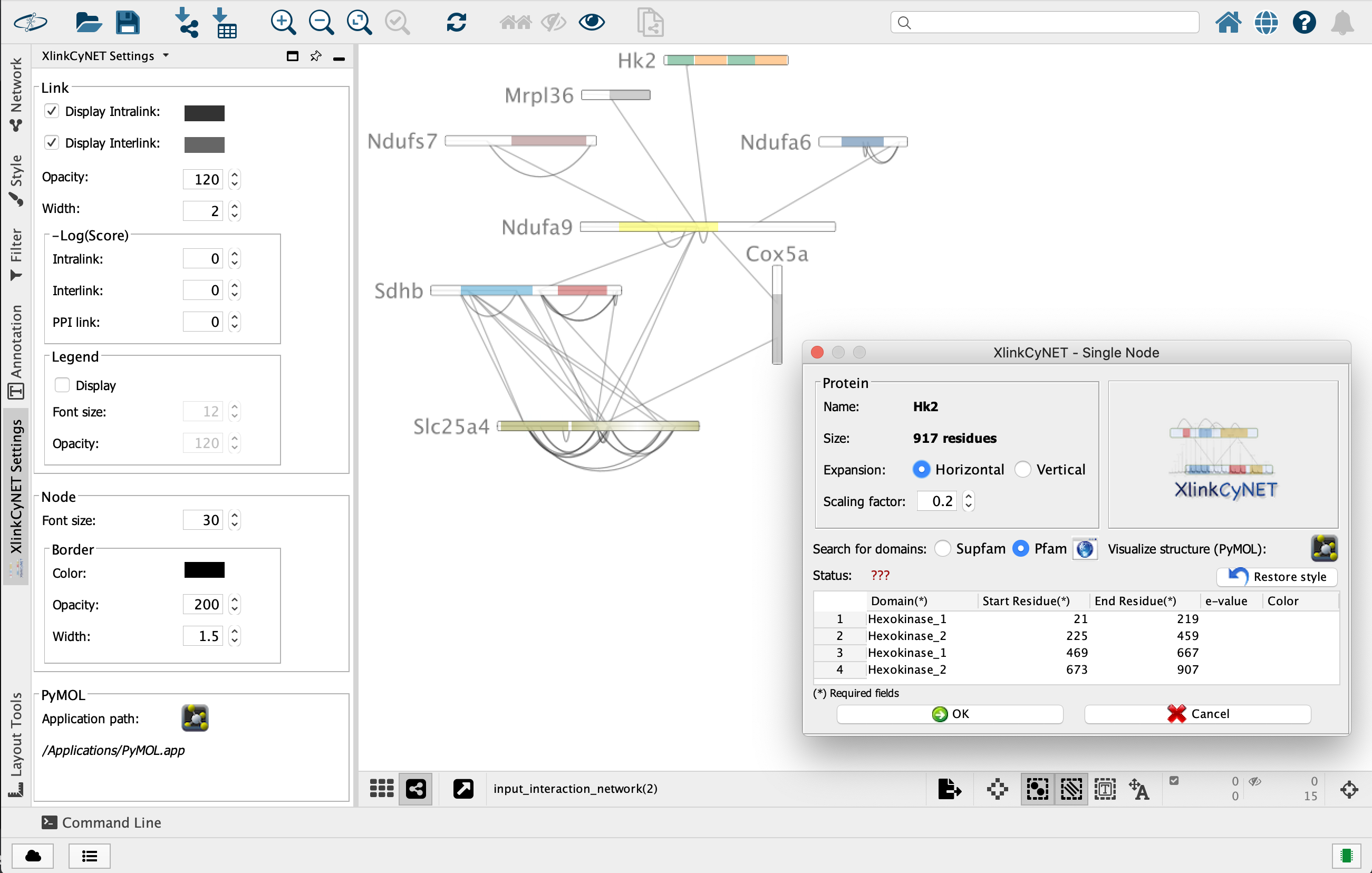
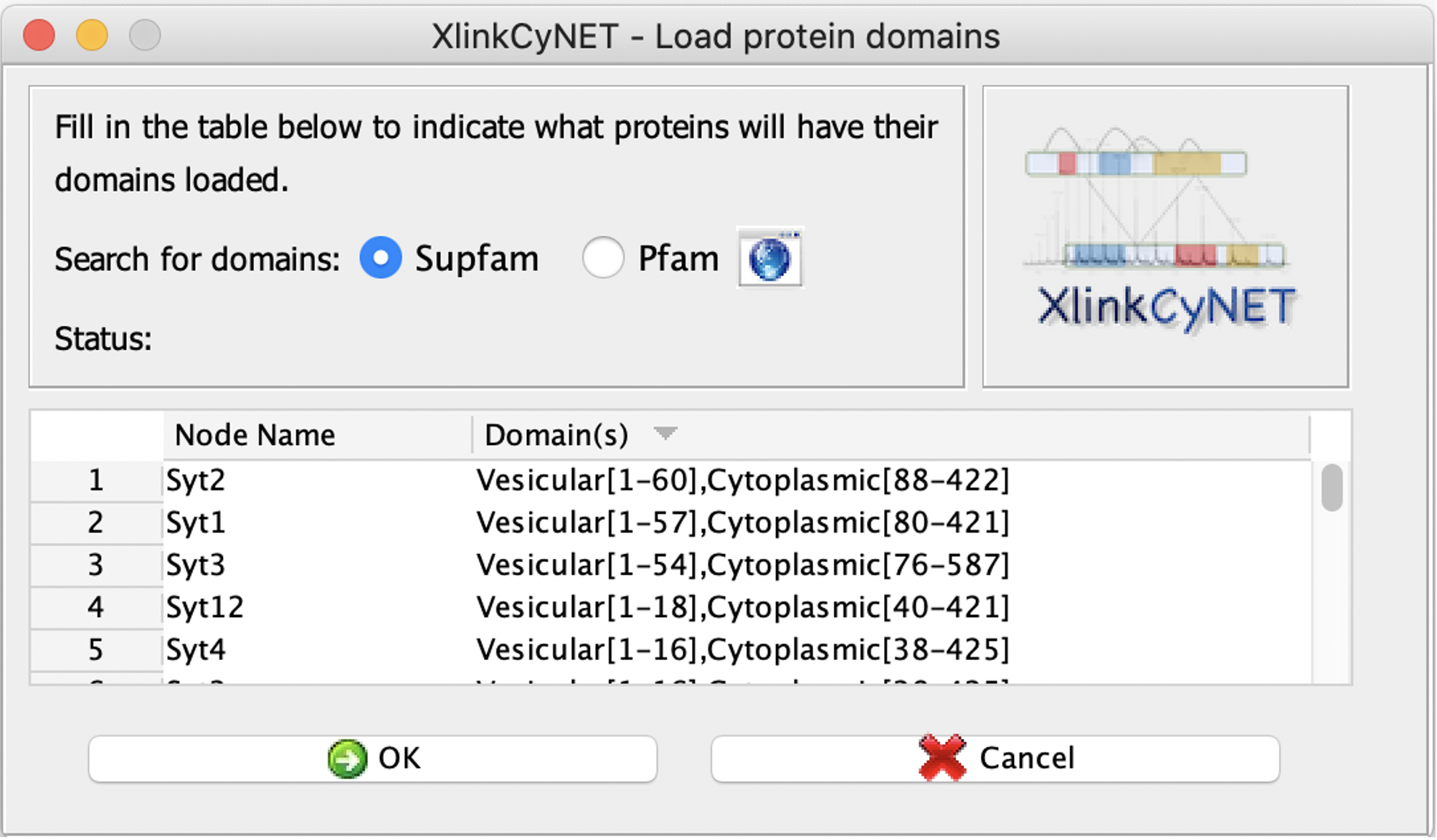
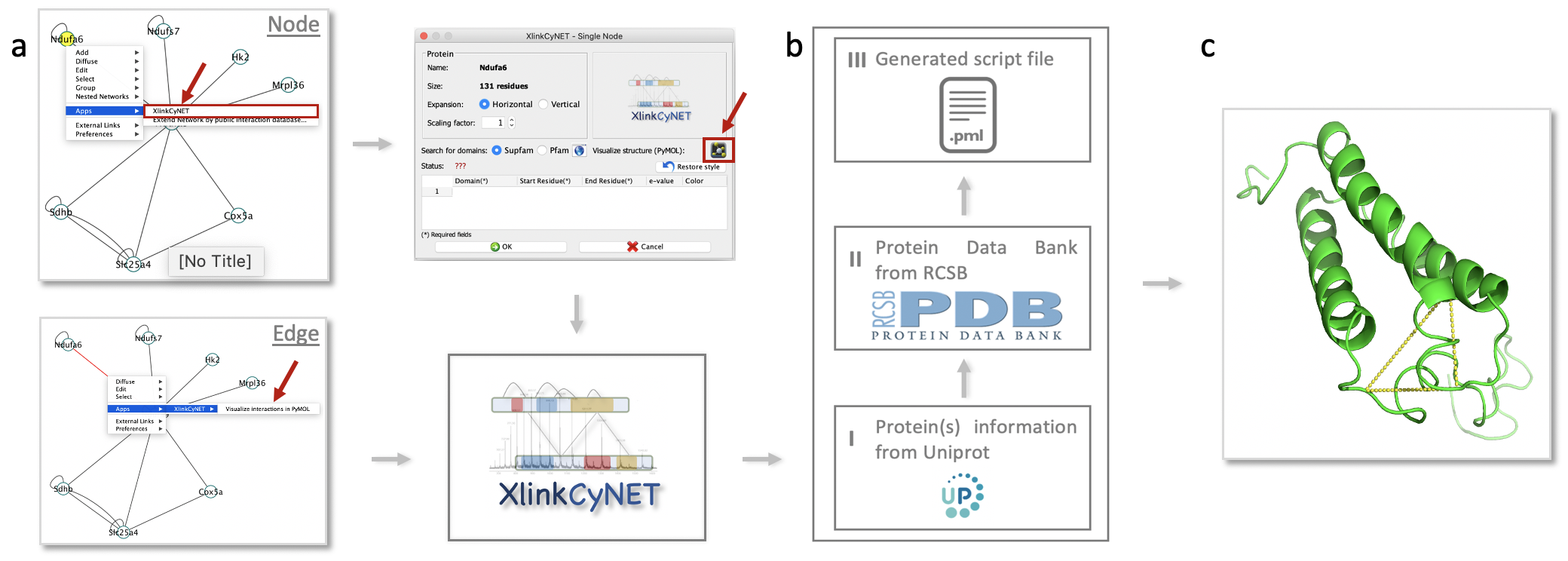
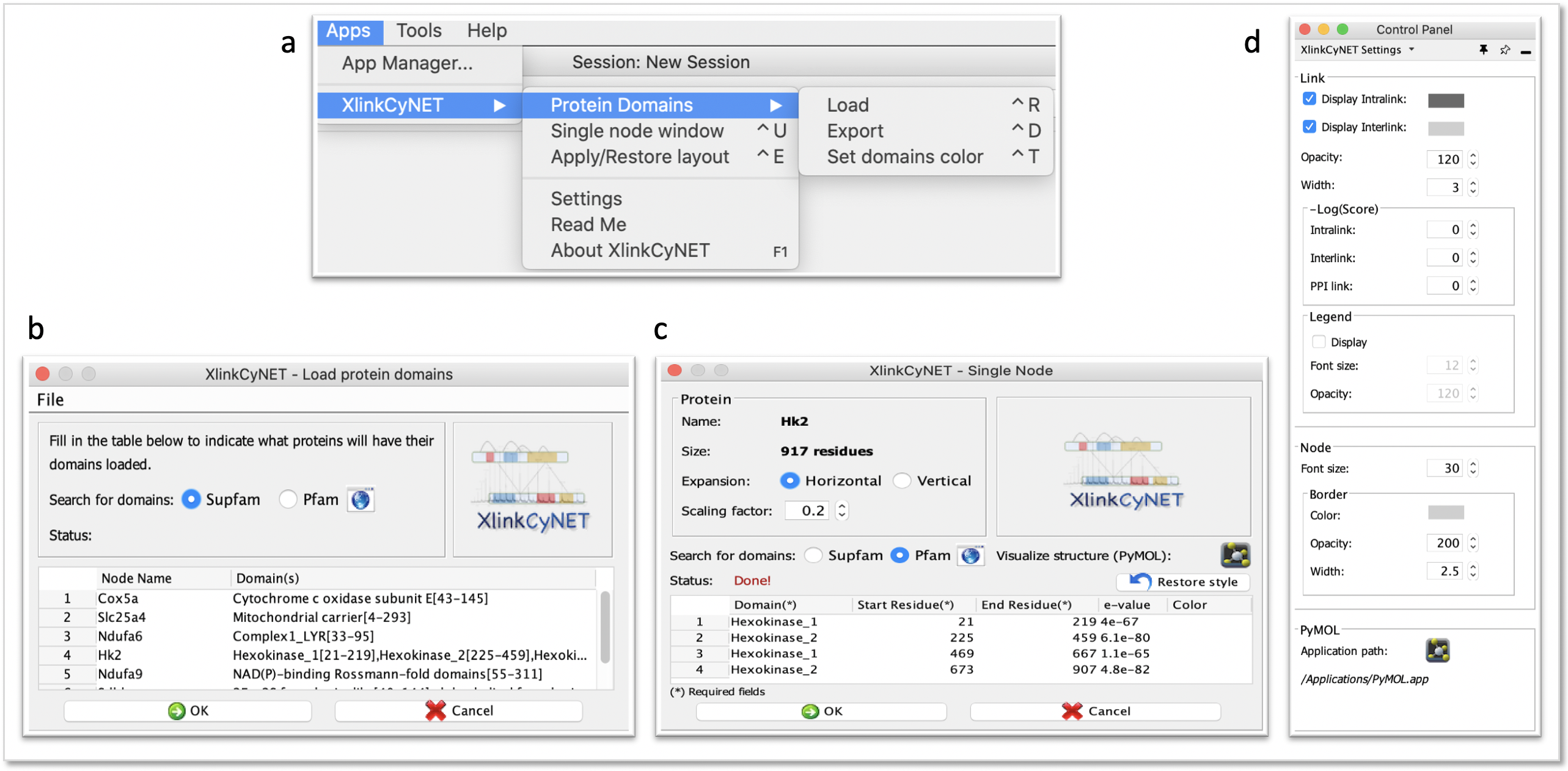
**XlinkCyNET** is an open-source Java plugin that generates residue-to-residue connections provided by XL-MS in protein interaction networks. Importantly, it provides an interactive interface for the exploration of cross-links and offers various options to display protein domains. Moreover, XlinkCyNET is integrated to ___PyMOL___, allowing to visualize all identified cross-links in the protein structure. The app works well in complex networks containing thousands of proteins.
A [video](https://www.youtube.com/embed/uLuJ7RlFb3I?rel=0&showinfo=0) demonstrating how the application works can be found on the [software website](https://www.theliulab.com/software/xlinkcynet).<br/>
A detailed [tutorial](http://dx.doi.org/10.21203/rs.3.pex-1172/v1), which explains step by step the functionality of all parameters is available at *Protocol Exchange*.
***Please cite our paper:***<br/>
Lima, DB, *et al.*, **[“XlinkCyNET: A Cytoscape Application for Visualization of Protein Interaction Networks Based on Cross-Linking Mass Spectrometry Identifications”](https://doi.org/10.1021/acs.jproteome.0c00957)**, Journal of Proteome Research (20): 1943–1950, 2021.
###Features
↪ Visualize protein-protein interaction network based on cross-linking mass spectrometry data<br/>
↪ Apply a residue-to-residue layout to a Cytoscape interaction network
↪ Annotate protein domains to all nodes in the network
↪ Map cross-links on high-resolution structures through PyMOL
↪ *AlphaFold* is possible to be used to retrieve protein structures.
<hr/>
###Input file
<p>The input file must have the following columns, which should be assigned to the correct column type in Cytoscape.</p>
*Required columns:*
`gene_a` → represents the node name: *source node (Local)*,
`gene_b` → represents the node name: *target node (Local)*,
`protein_a` → represents the Uniprot protein accession number of protein a in the following format: db|Unique_Identifier|Entry_Name: *Source Node Attribute (Shared)*,
`protein_b` → represents the Uniprot protein accession number of protein b in the following format: db|Unique_Identifier|Entry_Name: *Target Node Attribute (Shared)*,
`length_protein_a` → represents the number of residues of protein a: *Source Node Attribute (Shared)*,
`length_protein_b` → represents the number of residues of protein b: *Target Node Attribute (Shared)*,
`crosslinks_ab` → represents all links (inter and intra) between protein *a* and *b*. The links must be separated by ‘*#*’: *Edge attribute (Shared)*,
`crosslinks_ba` → represents all links (inter and intra) between protein *b* and *a*. The links must be separated by ‘*#*’: *Edge attribute (Shared)*,
*Optional columns:*
`ppi_score` → represents the score of the interaction between two proteins: *Edge attribute (Shared)*,
`score_ab` → represents the score of each intra/interlink from protein *a* to protein *b*. The value must be separated by '#': *Edge attribute (Shared)*. *Example: 2.33e-24#3.67e-19#8.55e-7*,
`score_ba` → represents the score of each intra/interlink from protein *b* to protein *a*. The value must be separated by '#': *Edge attribute (Shared)*. *Example: 2.33e-24#3.67e-19#8.55e-7*.
Example file can be found [here](https://github.com/diogobor/XlinkCyNET/tree/master/example_data).
<hr>
###Automation
↪ XlinkCyNET supports Cytoscape Automation from both commands and CyREST by providing six different commands: <br/>
<li>`applyRestoreStyle` <br/>attributes: <br/>
→ *nodesName*: Give the node(s) name, separated by comma, that will be expanded or restored. (type 'all' to set style to all nodes) <br/>
→ *proteinHorizontalExpansion*: Set whether the protein will be expanded horizontally or not. <br/>
→ *proteinScalingFactor*: Set a scale factor (between 0 and 1) to the protein length.</li><br/>
<li>`loadProteinDomains`<br/>attributes: <br/>
→ *fileName* : Name of the protein domain file. (Supported formats: .tab and .csv)<br/>→ *fromPfamServer*: Set which server the protein domains will be retrieved. (true = Pfam, false = Supfam)<br/>
→ *nodesName*: Give the node(s) name, separated by comma, to get domains. (type 'all' to get domains of all nodes)</li><br/>
<li>`exportProteinDomains`<br/>attribute: <br/>
→ *fileName*: Name of the protein domain file. (Supported format: *.csv)</li><br/>
<li>`setProteinDomainsColor`<br/>attributes: <br/>
→ *domainName*: Name of the protein domain to change the color.<br/>
→ *proteinDomainColor*: Set a color to a protein domain (*e.g., 'red' or '#FF0000' or 'FF0000'*)</li><br/>
<li>`loadPTMs `<br/>attributes: <br/>
→ *fileName* : Name of the PTM(s) file. (Supported formats: .csv)<br/>
→ *nodesName*: Give the node(s) name, separated by comma, to find PTM(s). (type 'all' to get domains of all nodes)</li><br/>
<li>`exportPTMs`<br/>attribute: <br/>
→ *fileName*: Name of the protein domain file. (Supported format: *.csv)</li><br/>
<li>`loadMonolinks `<br/>attributes: <br/>
→ *fileName* : Name of the Monolinked peptide(s) file. (Supported formats: .csv)</li><br/>
<li>`exportMonolinks`<br/>attribute: <br/>
→ *fileName*: Name of the protein domain file. (Supported format: *.csv)</li><br/>
<li>`setParameters`<br/>attributes: <br/>
→ *displayInterlinks*: Display or hide all identified interlinks<br/>
→ *displayIntralinks*: Display or hide all identified intralinks<br/>
→ *displayMonolinks*: Display or hide all identified intralinked peptides<br/>
→ *displayPTM*: Display or hide all PTM(s)<br/>
→ *displayLinksLegend*: Display or hide the legends of all identified cross-links<br/>
→ *fontSizeLinksLegend*: Set the font size of the legend of all identified cross-links<br/>
→ *fontSizeNodesName*: Set the font size of the name of all nodes<br/>
→ *interlinksColor*: Set a color to all identified interlinks (*e.g., 'red' or '#FF0000' or 'FF0000'*)<br/>
→ *intralinksColor*: Set a color to all identified intralinks (*e.g., 'red' or '#FF0000' or 'FF0000'*)<br/>
→ *pTMColor*: Set a color to all post-translational modifications (*e.g., 'red' or '#FF0000' or 'FF0000'*)<br/>
→ *monoLinksPeptideColor*: Set a color to all identified intralinked peptides (*e.g., 'red' or '#FF0000' or 'FF0000'*)<br/>
→ *nodeBorderColor*: Set a color to all nodes borders (*e.g., 'red' or '#FF0000' or 'FF0000'*)<br/>
→ *opacityBorderNodes*: Set the opacity of the border of all nodes (range between 0 - transparent and 255 - opaque)<br/>
→ *opacityLinks*: Set the opacity of all identified cross-links (range between 0 - transparent and 255 - opaque)<br/>
→ *opacityLinksLegend*: Set the opacity of the legend of all identified cross-links (range between 0 - transparent and 255 - opaque)<br/>
→ *scoreInterlink*: Set the threshold score to interlinks. All interlinks that have a -log(score) above the threshold will be displayed.<br/>
→ *scoreIntralink*: Set the threshold score to intralinks. All intralinks that have a -log(score) above the threshold will be displayed.<br/>
→ *scorePPIlink*: Set the threshold score to PPI links. All PPI links that have a -log(score) above the threshold will be displayed.<br/>
→ *widthBorderNodes*: Set the width of the border of all nodes (range between 1 and 10)<br/>
→ *widthLinks*: Set the width of all identified cross-links (range between 1 and 10)
</li><br/>
<li>`readMe`</li>
<hr/>
<p>Don't hesitate to send us any comments, questions or bug reports to: <img src="http://diogobor.droppages.com/emailBorderless.png" alt="email"/></p>
Any feedback is welcomed!<br/><br/>
Have fun! :)