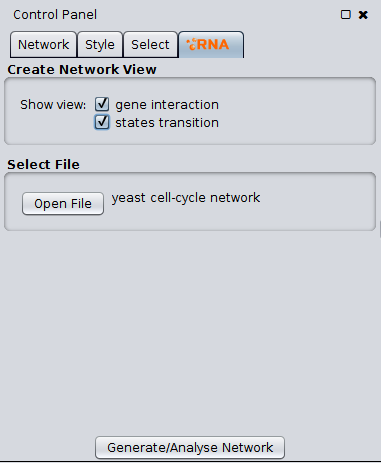
RNA takes as entry an interaction network, represented by a file of **.txt** format.
That file must contain a matrix of size **n** x **n**, where **n** is the number of genes/nodes.
The entry file shoud follow the notation:
- The first row contain the names of the genes, separated by comma (,);
-The following lines contain one of the following:
1: when the gene j is activated by the gene i;
0: when there is no interaction between i and j;
-1: when the gene j is inhibited by gene i;
- The numbers are also separated by comma (,).
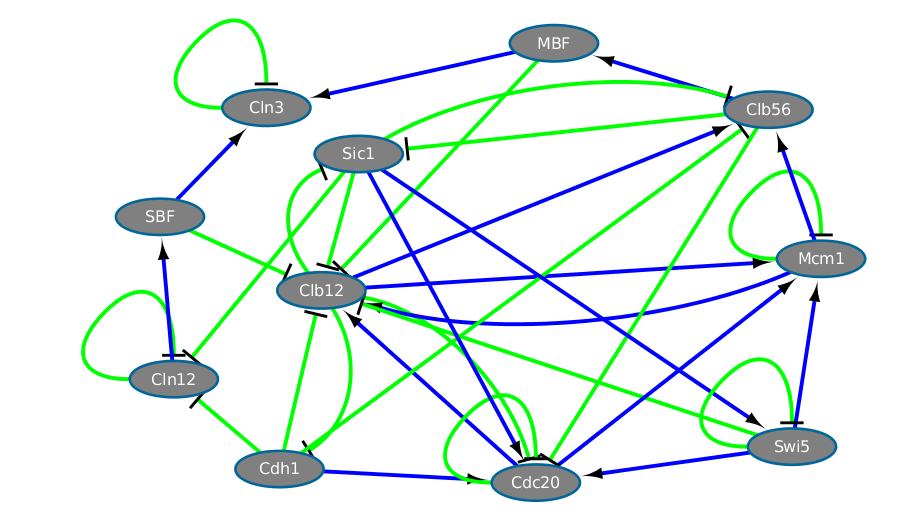
Here goes an example of matrix of the **yeast cell-cycle network**:
Cln3,MBF,SBF,Cln12,Cdh1,Swi5,Cdc20,Clb56,Sic1,Clb12,Mcm1
-1,0,0,0,0,0,0,0,0,0,0
1,0,0,0,0,0,0,0,0,-1,0
1,0,0,0,0,0,0,0,0,-1,0
0,0,1,-1,0,0,0,0,0,0,0
0,0,0,-1,0,0,1,-1,0,-1,0
0,0,0,0,0,-1,1,0,0,-1,1
0,0,0,0,0,0,-1,0,0,1,1
0,1,0,0,0,0,-1,0,-1,0,0
0,0,0,-1,0,1,1,-1,0,-1,0
0,0,0,0,-1,0,-1,1,-1,0,1
0,0,0,0,0,0,0,1,0,1,-1
Using the entry file, we generate the matrix visualization as a cytoscape network.
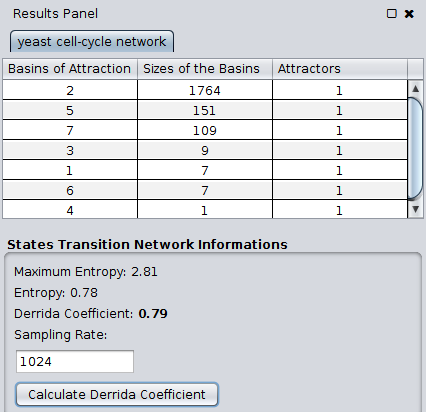
Along with that visualization we also generate a state transition network, that contains 2^n nodes, *so is possible that cytoscape will crash for regulatory networks with more than 20 nodes* (2^20), the layout of the network also affects the performance, by default the layout selected is *Grid Layout*, the grid layout was selected because of some tests of performance made. Each basin of attraction of the network has a different color.
The app also computes some information from the states transition network like the **entropy** and the **derrida coefficient**. These informations are shown in the results panel.
###Bug Reports
If you find a bug, please file a report at [https://github.com/Sibelly/rna-app/issues GitHub Issues].